|
���潭1����С��1�� ���п�2������2��������2��������1
��1�����з�װ�����о������������ص�ʵ���ң�����100029��
2������װѧԺ���Ͽ�ѧ�빤��ѧԺ������100029��
ժ Ҫ�����Ȼ���Ϊ��ϼ�����������6��PA6����ĩ���Ȼ����ܽ��ڼ���/�ȷ»����Һ���Ƶö�����Ʒ������DTA��XRD��FTIR�ȷ����Զ����ṹ�����ܽ��з�����̽����PA6Ũ�ȶԶ�����Ʒ�ṹ�����ܵ�Ӱ�졣����������Ȼ��ƿ��Բ�������PA6�������������ᾧ����������PA6Ũ�ȵ���������Ʒ�ķ��Ӽ���������ǿ���۵����ߣ���ͬŨ�ȼ�Ķ�����Ʒ�ṹ�������Լ���ϼ������Ч����һ���IJ��졣
�ؼ��ʣ�PA6���Ȼ��ƣ���ϣ�����
�Դ��ö�����˿���ɹ��Ʊ�����ǿ��ģ��UHMWPE��ά[1]���������Ƕ��������Դ������۱�ϩ��[2]������ϩ��[3]�����˴������о������Ͽ��������͵ĸ�������ά����֬���������6�������Ʊ��붳����˿���棬������Ҳ�����˺ܶ���о�����������û��̫���չ[4-5]��������Ϊ���Ӹ߶�ȡ�����Ʊ���ǿ��ģ��������ά�����Ĺؼ��������������Ӽ��ǿ�����������Լ�˷��ӵ�ȡ�����ά�ĸ߱����졣Ҫ�Ʊ������ܵľ�������ά������Ҫ�Դ���ӽ�����ϣ��ƻ�������������������á��ڿ�����ǰ�ڵĹ����У����������鿭�����˶�PA6/���Ȼ�����ϵ�����˳�����̽�����Ƶ��˸���ϵ�Ķ�����Һ���õ��˲�״�ṹ�ḻ�Ķ�������Ϊ����ϵ��Ҫ��ͨ����λ���γɽ�����ģ�ͬʱ�Զ���ԡ������һ����̽��[6-7]���Ʊ�����о����������������Ȼ�ﮡ���������������ϼ��ڲ�ͬ�ܼ������������ǿ��[8-9]���������Լ���/�ȷ�Ϊ����ܼ���CaCl2Ϊ��ϼ����Ʊ����������PA6/CaCl2������ϵ����ϸ������PA6Ũ�ȶԶ����Ľṹ�����ܵ�Ӱ�졣
1 ʵ�鲿��
1.1 ԭ����
UHMWPA6��ĩ�������������ٽ��ۺϷ��Ʊ������ճ����r="8.62��������װѧԺ���ƣ����ᡢ�ȷ¾�Ϊ��ѧ�������ۣ���ˮ�Ȼ��ƣ���ѧ�������ۡ�
1.2 PA6�������Ʊ�
�ڵ��������£���CaCl2�ܽ��ڼ������ȷ£������75/25���Ļ��Һ�С���CaCl2��ȫ�ܽ��������PA6��ĩ���ڵ��������³������͡������ܽ⣬��ֹ���ݣ��Ƶ�PA6/CaCl2/����/�ȷ¶�����ϵ�����У�CaCl2��PA6��Ħ���Ⱦ�Ϊ0.2��PA6��Ũ�ȣ�����������Ϊ20%��32%��60%������ʹ���Һ�߱������ԣ����䵹�ڲ���Ƭ�ϲ���ƽ����140���¸���2h����ȥ���ܼ���Ĥ�������ܲ��ԡ�
1.3 ��������
�ܽ�-����ת���¶ȣ�Tf������һ������PA6���Һ�����Թ��ڣ��ڱ�ԡ�б���24 h����ö������������Թܿڵ��ã�Ȼ����12��/h���������£����������䵽�Թ��²�ʱ���¶ȶ�ΪTf�����С��0.2��[10]��
�����ܣ����ò��ȷ�������DTA���������¶�40��280���������ٶ�15��/min��ͨN2����50ml/min����������Ϊ�α��
�ᾧ�ṹ�������ձ�Rigaku��˾��D/MAX-��A���X�������䣨WAXD�����ԣ�ͭ��Ni���ˣ��ܵ�ѹ40kV���ܵ���50mA��2��ɨ�跶Χ6��~36����ɨ���ٶ�Ϊ2�㡤min-1��
�������������������Nicolet��˾��NEXUS��FT-IR����Ҷ��������ǣ�ATR������ɨ�����64�Ρ�
2 ���������
2.1 �ܽ�-����ת���¶�
��1 ��ͬPA6Ũ�ȵ�PA6/CaCl2�������ܽ�-����ת���¶�
|
PA6Ũ��/% |
20% |
32% |
60% |
|
Tf/�� |
31.8 |
41.3 |
67 |
�ܽ�-����ת���¶ȵIJⶨ�����ǽ����ڽ�����¶ȿ����ܽ�-����ת�������ܽ�/�����Ľ��ޣ����ת�����������ҺŨ�ȡ��¶ȡ���Ӧʱ�����������ܶ��йء���Һͨ�����»��������������һ������¶ȷ�ת�����¶ȱ仯ʹ�γɵĶ����ٻָ����ܽ��������ܽ�-�����ȿ�����ת�䣩���������¶Ⱥ������¶����ֳ�ij�̶ֳȵIJ���Թܵ������Dzⶨ��Һ�Ķ������Լ��������ڵ�����ķ�������1�����˲�ͬPA6Ũ�ȵĶ�����Ʒ���ܽ�-����ת���¶�Tf����PA6��Ũ����20%����60%ʱ��Tf��31.8������67������һ�����PA6/GaCl3������ϵ����[7]��������ΪPA6Ũ��Խ��λ����ڵķ�������ĿԽ�࣬���γɶ����ķ��Ӽ�������ҲԽ������ʹ�����������������Խ�࣬����Ʒ��TfԽ�ߡ�
2.2 ������
ȡ����Һ������Ĥ��DTA��ͼ1��ͼ2�ֱ�Ϊ��ͬŨ�ȵ�PA6/CaCl2������ϵ�Լ���PA6��DTA��ͼ����PA6��Ʒ��222���ۻ������¹�������198���ᾧ���봿PA6��ȣ�PA6/CaCl2������Ʒ���۵������½�������ﵽ��150������������Ca2+����������ϣ������˲��������ʹ�ö�����Ʒ�Ľᾧ�ȼ�С��ֵ��ע����ǣ��������������õ�PA6���������ܼ������ͬ��CaCl2��PA6���Ħ����Ҳ��ͬ��PA6�ᾧ��Ҫ�Ƿ��Ӽ������ɣ�������������ͬ������£������Ͻ���ϼ�����������ı���Ӧ��Ҳһ�����ɴ������������Ľᾧ��Ӧ�ýӽ�����ʵ���ϣ�PA6Ũ�ȴ�20%����60%ʱ��������Ʒ���۵���70.1����ߵ�121.2����˵����ͬPA6Ũ�ȵĶ������нᾧ���Dz�ͬ�ġ�����ԭ�����£���1��PA6Ũ�����ӣ�CaCl2Ũ����Ӧ���ӣ����²����Ȼ��ƴ���Һ��������������Ϸ�Ӧ�����PA6����ϳ̶��½�����2��PA6Ũ�����ӣ���Һ�д�������������С��������������������������ϼ������뵽�������䣬ʹ��PA6������γ̶ȼ�С���ᾧ�������۵����ߡ�

ͼ1 ��ͬPA6Ũ�ȵ�PA6/CaCl2������ϵ��DTA����
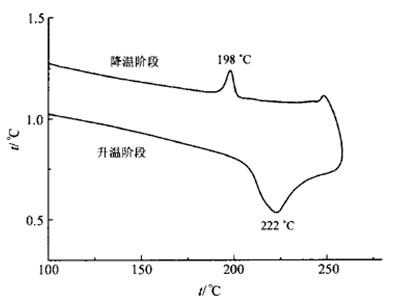
ͼ2 ��PA6��DTA����
2.3 XRD����
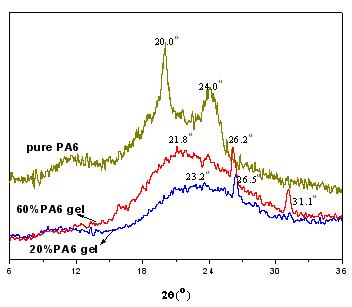
ͼ3��PA6��PA6/CaCl2������ϵ��XRDͼ
�����ﴦ����Ķ���Ĥ��XRD���ԣ�ͼ3�����˴�PA6��PA6/CaCl2������ϵ��XRD�Ա���ͼ����PA6�����ֱ��ڵ����������Ϊ 2��Ϊ20����24o�������������Ե�����壬Ϊ�����͵���������壬�þ��Ͷ�Ӧ���ڲ��ṹ��PA6����������ƽ�����У����Ӽ��γɵ������ࡣ����һ������CaCl2����������ʧ��ȡ����֮����һ����Բ�����ͷ壬˵��PA6�Ľᾧ����������������Ϊ��������PA6�������еIJ�������������������ã��ƻ����������ŵ�C="O��N-H֮���γɵ�������Ӷ�ʹ�ⲿ���������ᾧ��ͬʱ����Ĥ��XRD������26����������С������ķ壬�����������PA6�ᾧ�����Ƶ��´���ȱ���������¡����Ŷ�����PA6Ũ�ȵ�������Ĥ������ķ�λ��С�Ƕȷ���ƫ�ƣ���ǿ���ӣ����α�ü���˵��������PA6�Ľᾧ����������������и��ӹ�������20%�Ķ���Ĥ��Ʒ��ȣ�60%�Ķ���Ĥ��Ʒ��31.1���������Ƽ����С�壬�Ʋ�ΪCaCl2����������塣ʵ���У�CaCl2��PA6��Ħ���Ⱦ�Ϊ0.15������PA6Ũ�ȵ����ӣ�CaCl2��Ũ��Ҳ�����ӣ���140������Ĥʱ�ܼ��ӷ�������CaCl2����������������Ӷ���XRD��ͼ��������ӳ������DTA�IJ��Խ��һ�¡�
2.4 �������
ȡ����Һ������Ĥ��FT-IR���ԣ��õ���ͬPA6Ũ�ȵĶ������ĺ�����ͼ����ͼ4��ͼ��ͬʱ�����˴�PA6�Լ�PA6�ļ�����Һ���¸�����Ĥ��Ʒ�ĺ�����ͼ�������Աȡ���2�����˲�ͬ��Ʒ�и����ŵ�������Ƶ�ʡ�PA6������ҺĤ�и����ŵ������շ��λ���봿PA6�ļ���һ����û�м����C="O�������շ���֡�������Ϊ�������ﴦ�������Ἰ��ȫ��ȥ�������������Ʒ������������������������6��Ʒ�����PA6/CaCl2������Ʒ��N-H����������3299cm-1�ƶ���3246m-1�������ϴ�����ƣ�����I����C="O��������1638 cm-1�ƶ���1631cm-1���������ƣ�����II����C�CN��������1543 cm-1�ƶ���1562cm-1�������ϴ�ĺ��ơ���������������յ����ø�ǿ�ĸ�����ȡ��������������ŵ�C="O����������ã�������N�CH��C="O���������Ӷ�ʹ���ߵ����������շ���Ͳ����ƶ���ͬʱ��������������ɶ�C="O�и�ǿ���յ����ã�ʹ��O��H�����ʻ���p-���������ü�����������C�CN��������ʹ���������շ��������ƶ�[11]���Ա�����Ũ�Ȳ�ͬ��PA6������Ʒ�ĺ�����ͼ���Կ�����Ũ�ȶԸ����ŵ������շ�λ�ü���û��Ӱ�죬ֻ��ǿ��������ͬ�������������Ʒ���������ŵ���϶Ȳ�ͬ�йء����⣬����������Ʒ����1712cm-1������һ���������շ壬�Ʋ�Ϊ������C="O�������壬������Ƕ�����Ʒδ�����¸��ﴦ���������������������¡�

ͼ4��PA6��PA6������Ĥ��PA6/CaCl2����Ĥ�ĺ�����ͼ
���У�a-��PA6��b-20%PA6 gel��c-60%PA6 gel��d- PA6������ҺĤ
��2��PA6��PA6������Ĥ��PA6/CaCl2����Ĥ�ĺ�����ͼ�и����ŵ�������Ƶ��
|
��Ʒ |
��N�CH |
��C="O |
��sCH2 |
��asCH2 |
��C�CN |
|
��PA6 |
3299 |
1638 |
2865 |
2939 |
1543 |
|
20%PA6 gel |
3246 |
1631 |
2864 |
2931 |
1562 |
|
60%PA6 gel |
3245 |
1631 |
2864 |
2933 |
1562 |
|
PA6������ҺĤ |
3295 |
1639 |
2864 |
2932 |
1544 |
3 ����
�Ȼ��ƿ�����PA6����������λ���������������PA6����ӵĽᾧ����������PA6Ũ�ȵ���������ϵ�PA6/�Ȼ��ƶ�����Ʒ�д���Ӽ�������������ܽ�-����ת������ߣ��۵�����PA6Ũ��̫��ʱ���������е��Ȼ��ƻ�����������ȫ������Ϸ�Ӧ������ʵ����϶ȵ��½���
�����
[1] Smith P, Lemstra P J, Booij H C. Ultradrawing of high-molecular-weight polyethylene cast from solution. II. Influence of initial polymer concentration[J]. J Polym Sci: Polym Phys Ed, 1981, 19: 877
[2] Chaea H G, Minusa M L, Rasheeda A, Kumar S. Stabilization and carbonization of gel spun polyacrylonitrile/single wall carbon nanotube composite fibers[J]. Polymer, 2007, 48: 3781
[3] Yamaura K, Kumakura R. Gel-spinning of partially saponificated poly(vinyl alcohol)[J]. J Appl Polym Sci, 2000, 77: 2872
[4] Matsuo M, Sato R, Yanagida N, Shimizu Y. Deformation mechanism of nylon 6 gel and melt films estimated in terms of orientation distribution function of crystallites[J]. Polymer, 1992, 33:1640
[5] Cho J W, Lee G W, Chun B C. Mechanical properties of Nylon-6 fibers gel-spun from benzyl alcohol solution[J]. J Appl Polym Sci, 1996, 62: 771
[6] ������, ��С��, ���п���. ����6�����������[J]. ������װѧԺѧ��, 2006, 26(4): 1
[7] �鿭�ɣ���С�������п���. �����ճ�Ⱦ�����6�������о�[J]. �ϳ���ά��ҵ, 2008,31(1): 15
[8] �Ʊ�, ��С��, ����. ����6�ֲ���ϵ��о�[J]. ������װѧԺѧ��, 2008, 28(2): 2
[9] ţ��, ��С��, ���п�. ��϶Ը�ճ������6���ܵ�Ӱ��[J]. ������װѧԺѧ��, 2006, 26(4): 7
[10] Eldridge JE, Ferry JD. Studies of the cross-linking process in gelatin gels III. Dependence of melting point on concentration and molecular weight[J]. J Phys Chem, 1954, 58(11): 992
[11] ����,�����,�Ű���.�Ȼ��ƶ�����6 ����ת����̵���λ�����о�[J]. ����ѧ�������,2004,24(3):295
|








